
Definición
Es un trastorno sanguíneo que se transmite de padres a hijos (hereditario) en el cual el cuerpo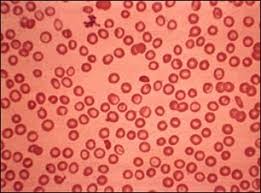 produce una forma anormal de hemoglobina, la proteína en los glóbulos rojos que transporta el oxígeno. Este trastorno ocasiona destrucción excesiva de los glóbulos rojos y anemia.
produce una forma anormal de hemoglobina, la proteína en los glóbulos rojos que transporta el oxígeno. Este trastorno ocasiona destrucción excesiva de los glóbulos rojos y anemia.
Síntomas
El defecto o deleción de un gen en la talasemia β causa una anemia hemolítica que oscila entre leve y moderada sin síntoma alguno. La deleción de dos genes ocasionan anemia más severa y la presencia de síntomas: debilidad, fatiga, dificultad respiratoria. En las variantes más graves, como la talasemia beta mayor, pueden aparecer ictericia, úlceras cutáneas, cálculos biliares y agrandamiento del bazo (que en ocasiones llega a ser enorme). La actividad excesiva de la médula ósea puede causar el ensanchamiento y el agrandamiento de algunos huesos, especialmente los de la cabeza y del rostro.
Los huesos largos tienden a debilitarse y fracturarse con gran facilidad. Los niños que padecen ciertas talasemias pueden crecer con más lentitud y llegar a la pubertad más tarde de lo normal. Como la absorción del hierro puede aumentar c
omo respuesta a la anemia sumado al requerimiento de transfusiones de sangre frecuentes (las cuales suministran más hierro), es posible que se acumulen cantidades excesivas de hierro y se depositen en la musculatura del corazón, causando insuficiencia cardíaca.
Las talasemias son más difíciles de diagnosticar que otros trastornos de la hemoglobina. El análisis de una gota de sangre por electroforesis puede ser útil pero no concluyente, en especial en el caso de talasemia alfa. Por lo tanto, el diagnóstico se basa habitualmente en patrones hereditarios y en análisis especiales de hemoglobina. Por lo general, las personas que padecen talasemia no requieren tratamient
o alguno, pero aquellas con variantes graves pueden requerir un trasplante de médula ósea. La terapia con genes se encuentra en fase de investigación.
Clasificación
Los diferentes tipos de talasemia se clasifican según las cadenas de la hemoglobina cuya síntesis esté afectada:
Alfa - Talasemia
Es un déficit de síntesis de cadenas alfa de hemoglobina. Los síntomas varían en función del número de genes implicados en el trastorno.
- Si sólo está afectado un gen , los individuos permanecen asintomáticos, pero se transmite a la descendencia.
- Si se alteran dos genes: nos encontramos ante la talasemia minor . Los pacientes af
ectados pueden padecer anemias graves. En los recién nacidos pueden detectarse pequeñas cantidades de un tipo especial e inestable de hemoblobina, denominada de Barts, que desaparece pasado el primer mes de la vida.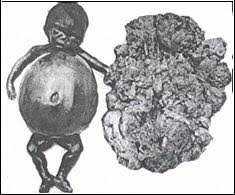
- Si sólo es normal un gen: se denomina enfermedad de la hemoglobina H , ya que conlleva a la formación de este tipo de Hb, que es muy inestable. El paciente presenta anemia severa, esplenomegalia, infecciones frecuentes y cálculos vesiculares.
- Si todos los genes están afectados: es la talasemia alfa mayor o síndrome del hydrops fetalis y provoca muerte intrauterina o a las pocas horas del nacimiento.
Beta -Talasemia
Los individuos heredan un gen de cada progenitor, lo que ofrece varias posibilidades de presentación de la enfermedad: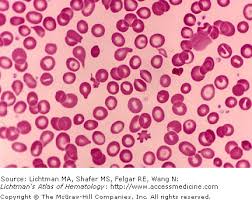
1.- Enfermedad de Cooley (homocigótica)
Existe una anemia severa. Los síntomas aparecen en los primeros meses de vida, donde los niños adquieren un tinte pálido, presentan picos febriles inesperados y abdomen distendido. La facies es mongoloide y radiológicamente existe aplanamiento de las costillas y malformación del cráneo y alteraciones óseas.
El tratamiento requiere supervisión hospitalaria permanente, esplenectomía si existe hiperesplenismo (aunque nunca antes de los 5 años), desferoxamina, vitamina E, transfusiones de concentrados de hematíes con el objeto de mantener la hemoglobina entre 10 - 12 gr/dl. Se deberá evitar el tratamiento con hierro, y si es posible, trasplante de médula ósea.
2.- Enfermedad de Rietti-Greppi-Micheli (heterocigótica)
Se denomina también rasgo talasémico , es un trastorno muy frecuente que no se acompaña de clínica. Raramente aparecen úlceras crurales o esplenomegalia. Existe una anemia moderada. No se debe administrar hierro a estos pacientes, salvo que sea realmente necesario ya que existe el peligro de producir depósitos patológicos del metal. En ocasiones es necesaria la administración de ácido fólico.
FUENTES
http://es.wikipedia.org/wiki/Beta_talasemia
http://images.google.com.mx/imgres?imgurl=http://www.mdconsult.com/das/patient/body/0/0/10041/1499_es.jpg&imgrefurl=http://www.mdconsult.com/das/patient/body/0/0/10041/31363.html&usg=__e3_mDYeQ7aAQb7VM3wBBY53slE4=&h=226&w=306&sz=19&hl=es&start=3&um=1&tbnid=ucilV8Ov8dYv5M:&tbnh=86&tbnw=117&prev=/images%3Fq%3DTALASEMIA%26hl%3Des%26sa%3DN%26um%3D1
.jpg)
Buenos dias
ResponderEliminarLas imagenes no se ven.
Sonia