*ALTERACIONES DE LOS GLOBULOS ROJOS
= El aspecto de los glóbulos rojos vistos al microscopio nos puede ofrecer diversas variables que orientan a diferentes enfermedades.
ALTERACIONES DEL TAMAÑO
LOS HEMATÍES NORMOCÍTICOS PRESENTAN LAS SIGUIENTES DIMENSIONES:
DIÁMETRO LONGITUDINAL: 7-8 Ð.
DIÁMETRO TRANSVERSAL PERIFÉRICO 2 Ð Y CENTRAL 1 ÐÐ
SUPERFICIE: 120-140 Ð2
VOLUMEN 80-100Ð3

ANISOCITOSIS
CONSISTE EN LA COEXISTENCIA, EN UNA MISMA MUESTRA DE SANGRE, DE HEMATÍES DE DISTINTOS TAMAÑOS. SE PRODUCE, POR EJEMPLO, EN LOS PACIENTES TRANSFUNDIDOS.

MICROCITOSIS
CONSISTE EN LA EXISTENCIA DE UNOS HEMATÍES CON UN DIÁMETRO LONGITUDINAL INFERIOR A 7 Ð Y UN VOLUMEN INFERIOR A 80 Ð3. SE PRODUCE EN LAS TALASEMIAS, EN LAS ANEMIAS SIDEROACRÉSTICAS Y, SOBRETODO, EN LAS ANEMIAS FERROPÉNICAS.

MACROCITOSIS
CONSISTE EN LA EXISTENCIA DE UNOS HEMATÍES CON UN DIÁMETRO LONGITUDINAL SUPERIOR A 8 Ð Y UN VOLUMEN SUPERIOR A 100 Ð3. SE PRODUCE EN EL ALCOHOLISMO Y EN LAS HEPATOPATÍAS CRÓNICAS.

MEGALOCITOSIS
CONSISTE EN LA EXISTENCIA DE UNOS HEMATÍES CON UN DIÁMETRO LONGITUDINAL SUPERIOR A 11 Ð. SE PRODUCE EN LAS ANEMIAS MEGALOBLÁSTICAS.

ALTERACIONES DE LA FORMA
ACANTOCITOSIS
CONSISTE EN LA EXISTENCIA DE UNOS HEMATÍES CON ESPECULAS DE LONGITUD Y POSICIÓN IRREGULAR (ACANTOCITOS). SE PRODUCE EN LA ABETALIPOPROTEINEMIA Y EN LA CIRROSIS HEPÁTICA.

DIANOCITOSIS
CONSISTE EN LA EXISTENCIA DE UNOS HEMATÍES PLANOS Y CON UNA FORMA DE SOMBRERO MEJICANO. ESTO HACE QUE LOS HEMATÍES, VISTOS FRONTALMENTE, TENGAN UN REBORDE COLORADO, QUE DELIMITA UNA ZONA ANULAR PÁLIDA, CUYO CENTRO TAMBIÉN ESTA COLORADO. ELLO LES CONFIERE UNA IMAGEN EN DIANA Y POR ESO, RECIBEN EL NOMBRE DE DIANOCITOS. LOS LEPTOCITOS SON HEMATÍES TAMBIÉN ANORMALMENTE DELGADOS PERO QUE NO ADOPTAN UN ASPECTO EN DIANA. SE PRODUCE EN TALASEMIAS Y EN LAS HEPATOPATÍAS.

DREPANOCITOSIS
CONSISTE EN LA EXISTENCIA DE UNOS HEMATÍES CON UNA FORMA DE HOZ. SE PRODUCE EN LA ANEMIA DE CÉLULAS FALCIFORMES.

ELIPTOCITOSIS
CONSISTE EN LA EXISTENCIA DE UNOS HEMATÍES CON UNA FORMA ELÍPTICA Y OVAL. SE PRODUCE EN LAS ANEMIAS FERROPÉNICAS, EN LAS ANEMIAS MEGALOBLÁSTICAS Y EN LA MIELOFIBROSIS, PERO ES TÍPICA DE LA ELIPTOCITOSIS HEREDITARIA. TIENEN ESTA FORMA LOS ERITROCITOS DEL CAMELLO, DE LA SALAMANDRA Y DE LA GALLINA.
 EQUINOCITOSIS
EQUINOCITOSISTAMBIÉN LLAMADOS ESTEREOCITOS O ASTROSITOS, CONSISTE EN LA EXISTENCIA DE UNOS HEMATÍES CON ESPECULAS CORTAS Y DISTRIBUIRLAS REGULARMENTE A LO LARGO DE TODA SU SUPERFICIE. SE PRODUCE, POR EJEMPLO, EN LA UREMIA, CUANDO LOS HEMATÍES SON POBRES EN K+ Y EN LAS HEPATOPATÍAS NEONATALES.

ESFEROCITOSIS
CONSISTE EN LA EXISTENCIA DE UNOS HEMATÍES CON UNA FORMA ESFÉRICA, QUE HABITUALMENTE TAMBIÉN SON DE PEQUEÑO (MICROESFEROCITOS). SE PRODUCE EN LA HIDROCITOSIS, EN LAS ANEMIAS HEMOLÍTICAS AUTOINMUNES Y, SOBRE TODO, EN LA ESFEROCITOSIS HEREDITARIA.

ESQUISTOCITOSIS
CONSISTE EN LA EXISTENCIA DE UNOS HEMATÍES FRAGMENTADOS. SE PRODUCE EN LA ANEMIA HEMOLÍTICA MICROANGIOPÁTICA, EN LA HEMÓLISIS MECÁNICA POR LA PRESENCIA DE UNA PRÓTESIS VALVULAR EN EL CORAZÓN Y EN LAS QUEMADURAS GRAVES.
 ESTOMATOCITOSIS
ESTOMATOCITOSISCONSISTE EN LA EXISTENCIA DE UNOS HEMATÍES CON UNA INVAGINACIÓN CENTRAL EN FORMA DE BOCA. ESTOS ERITROCITOS SON REALMENTE DISCOS UNICÓNCAVOS. SE PRODUCE EN EL ALCOHOLISMO Y EN LAS HEPATOPATÍAS CRÓNICAS.

EXCENTROCITOSIS
CONSISTE EN LA EXISTENCIA DE UNOS HEMATÍES CUYA HB ESTÁ CONCENTRADA EN UNO DE SUS POLOS. SE PRODUCE EN EL DÉFICIT DE GLUCOSA 6-FOSFATO DESHIDROGENASA.

KERATOCITOSIS
CONSISTE EN LA EXISTENCIA DE UNOS HEMATÍES CON DOS ESPECULAS EN SU SUPERFICIE. SE PRODUCE EN LA ANEMIA HEMOLÍTICA MICROANGIOPÁTICA, EN LA HEMÓLISIS POR PRÓTESIS CARDIACAS Y EN EL HEMANGIOMA CAVERNOSO.

POIQUILOCITOSIS
CONSISTE EN LA EXISTENCIA DE UNOS HEMATÍES CON UNA SOLA PROLONGACIÓN ALARGADA QUE LES CONFIERE EL ASPECTO DE UNA RAQUETA O DE LAGRIMA (DACRIOCITOS). SE PRODUCE EN LAS TALASEMIAS.

ALTERACIONES DEL COLOR:
LOS HEMATÍES NORMOCRÓMICOS PRESENTAN, CON LOS MÉTODOS DE TINCIÓN, HABITUALES, UNA COLORACIÓN ROSADA. ADEMÁS, ESTA COLORACIÓN ES MÁS INTENSA EN LA PERFIDIA QUE EN EL CENTRO.

ANISOCROMÍA
CONSISTE EN UNA FALTA DE UNIFORMIDAD EN LA COLORACIÓN ENTRE UNOS HEMATÍES Y OTROS. LA COEXISTENCIA DE DOS POBLACIONES DE HEMATÍES, CON COLORACIONES DISTINTAS, SE PRODUCE POR EJEMPLO EN:
EL INICIO DEL TRATAMIENTO DE LAS ANEMIAS CARENCIALES.
LOS ENFERMOS CON ANEMIA HIPOCROMA QUE SON TRASFUNDIDOS.

HIPOCROMÍA
CONSISTE EN LA EXISTENCIA DE UNOS HEMATÍES PÁLIDOS Y CON AUMENTO DE LA CLARIDAD CENTRAL (HEMATÍES HIPOCRÓMICOS Y ANULOCITOS). SE PRODUCE, POR EJEMPLO, EN LAS ANEMIAS FERROPÉNICAS.

HIPERCROMÍA
CONSISTE EN LA EXISTENCIA DE UNOS HEMATÍES INTENSAMENTE COLOREADOS (EMITES HIPERCRÓMICOS). SE PRODUCE EN LA ESFEROCITOSIS HEREDITARIA.

POLICROMASIACONSISTE EN LA EXISTENCIA DE UNOS HEMATÍES QUE PRESENTAN UNA COLORACIÓN LIGERAMENTE BASÓFILA. REALMENTE, ESTAS CÉLULAS SON RETICULOCITOS.

INCLUSIONES INTRAERITROCITARIAS
NORMALMENTE, LOS HEMATÍES SOLO CONTIENEN HB, POR LO QUE SU INTERIOR ES HOMOGÉNEO. SON EMBARGO, EN ALGUNAS OCASIONES SE PUEDEN ENCONTRAR ELEMENTOS EXTRAÑOS EN SU INTERIOR:
SUSTANCIA GRANULOFILAMENTOSA
LA SUSTANCIA GRANULOFILAMENTOSA O RELICULOFILAMENTOSA PROCEDE, FUNDAMENTALMENTE, DE RESTOS RIBOSÓMICOS AGREGADOS. CONSISTE EN UNA TRAMA GRANULOSA VISIBLE MEDIANTE LA COLORACIÓN CON AZUL DE CRESIL BRILLANTE. ES PROPIA DE LOS RETICULOCITOS.

CUERPOS DE HEINZ
SON PRECIPITADOS DE HB. CONSISTE EN UNA SERIE DE PEQUEÑAS GRANULACIONES QUE SE SITÚAN EN LA PERIFERIA DE LOS HEMATÍES Y QUE SE TIÑEN DE COLOR PÚRPURA CON UNA SOLUCIÓN DE CRISTAL VIOLETA. SE PRODUCEN EN ENFERMEDADES CONGÉNITAS QUE COMPORTAN UNA INESTABILIDAD DE LA HB, QUE HACE QUE ESTA SE DESNATURALICE Y PRECIPITE EN PRESENCIA DE ALGUNOS MEDICAMENTOS.

CUERPO DE HOWELL-JOLLY
ES UN PEQUEÑO RESIDUO NUCLEAR. CONSISTE EN UN GRUMO VISIBLE EN EL INTERIOR DE LOS HEMATÍES Y QUE SE TIÑE, DE UN COLOR QUE OSCILA ENTRE EL ROJO OSCURO Y EL NEGRO, CON LOS COLORANTES HABITUALES. APARECE EN SUJETOS ESPLENECTOMIZADOS Y EN LOS QUE PADECEN SPRUE.

CUERPOS DE PAPPENHEIMER
TAMBIÉN SE LLAMAN GRÁNULOS SIDERÓTICOS. SON ACÚMULOS DE HEMOSIDERINA UNIDA A PROTEÍNAS. CONSISTEN EN GRÁNULOS BASÓFILOS, CON LAS TINCIONES HABITUALES, QUE ADEMÁS, SE TIÑEN TAMBIÉN DE AZUL CON EL COLORANTE DE PERLS. SE PRODUCEN EN LOS ENFERMOS ESPLENECTOMIZADOS Y EN LAS ANEMIAS SIDEROACRÉSTICAS.

PUNTEADO BASÓFILO
PUEDEN SER AGREGADOS RIBOSÓMICOS ORIGINADOS POR UNA DEGENERACIÓN VACUOLAR DEL CITOPLASMA O PRECIPITADOS DE CADENAS GLOBÍNICAS LIBRES. CONSISTE EN PUNTITOS BASÓFILOS, CON LAS TINCIONES HABITUALES, DE TAMAÑO VARIABLE U DISPERSOS POR TODA LA SUPERFICIE DEL HEMATÍE. SE TIÑEN CON TINCIÓN DE PERLS. SE PRODUCE EN LAS INTOXICACIONES POR PLOMO, Y TAMBIÉN EN LAS TALASEMIAS Y EN LAS LEUCEMIAS.

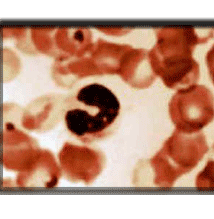
ANILLO DE CABOT
ESTÁN FORMADOS POR RESTOS DE LA MEMBRANA NUCLEAR O DE MICROTÚBULOS. CONSISTEN EN UNA ESPECIE DE HILOS BASÓFILOS, CON LAS TINCIONES HABITUALES, QUE ADOPTAN UNA FORMA DE ANILLO O DE OCHO Y QUE PUEDEN OCUPAR TODA LA PERIFERIA CELULAR. SE PRODUCE EN LAS ANEMIAS MEGALOBLÁSTICAS.DDD

Anillo cabot
INCLUSIONES PARASITARIAS
SON EJEMPLOS, LAS QUE SE ENCUENTRAN EN LOS HEMATÍES PARASITAZOS POR DISTINTAS FORMAS EVOLUTIVAS DEL PLASMODIUM. ASÍ PUES, SI EL HEMATÍE ESTA PARASITAZO POR UN TROFOZOITO INICIAL, LA IMAGEN PERCIBIDA DENTRO DEL HEMATÍE TIENE FORMA ANULAR (ANILLO PALÚDICO). EN LA BABESIOSIS TAMBIÉN SE PUEDEN OBSERVAR IMÁGENES, CON ASPECTO DE ANILLO, EN EL INTERIOR DE LOS HEMATÍES. SIN EMBARGO, EN ALGUNAS ESPECIES DE BABESIA, LOS TROFOZOITOS GENERAN IMÁGENES EN FORMA DE PERA EN EL INTERIOR DE LOS ERITROCITOS.

Anillo paludico
FUENTES:
http://html.rincondelvago.com/alteracion-de-los-hematies.html












 Existen sustancias que se oponen a la hemostasia como PGI2 o prostaciclina: proteina que inhibe la acción de proteinas activadas (factor de coagulación) antitrombina II: proteina circulante que bloquea los factores de la coagulación.
Existen sustancias que se oponen a la hemostasia como PGI2 o prostaciclina: proteina que inhibe la acción de proteinas activadas (factor de coagulación) antitrombina II: proteina circulante que bloquea los factores de la coagulación.














































.jpg)
{kind=link}
{kind=link}