
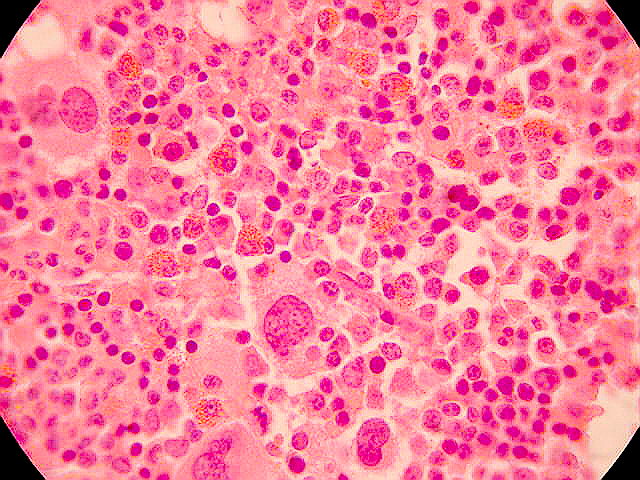
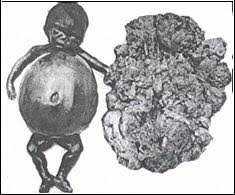
*LINFOMA *
Los linfomas son un conjunto de enfermedades cancerosas que se desarrollan en el sistema linfático, que también forman parte del sistema inmunológico del cuerpo humano. A los linfomas también se les llama los tumores sólidos hematológicos para diferenciarlos de las leucemias.

*CLASIFICACION DE LINFOMAS*
Linfomas precursores de células B: leucemia linfoblástica precursora aguda de células B (LLA-B, y linfoma linfoblástico precursor de células B (LBL, por sus siglas en inglés).
Linfomas periféricos de células B.
Leucemia linfocítica crónica de células B y linfoma linfocítico pequeño de células B.
Leucemia prolinfocítica de células B.
Linfoma/inmunocitoma linfoplasmacítico.
Linfoma de células de manto.
Linfoma folicular.
Linfoma extranodal de zona marginal de células B de tipo MALT.
Linfoma nodal de zona marginal de células B (de células B ± monocitoide).
Linfoma esplénico de zona marginal (linfocitos ± vellosos).
Leucemia de células pilosas.
Plasmacitoma y mieloma de células plasmáticas.
Linfoma de células B grandes difuso.
Linfoma de Burkitt.

Linfomas de células T y células Nk
Linfomas precursores de células T: leucemia linfoblástica precursora aguda de células T (LLA-T) y linfoma linfoblástico precursor de células T (LBL, por sus siglas en inglés).
Linfomas de células asesinas naturales (NK) y células T periféricas.
Leucemia linfocítica y leucemia prolinfocítica crónicas de células T.
Leucemia linfocítica granular de células T.
Micosis fungoide y el síndrome de Sézary.
Linfoma periférico de célula T, sin alguna otra caracterización.
Linfoma hepatoesplénico de células T gamma y delta.
Linfoma de apariencia paniculítica subcutáneo de células T.
Linfoma angioinmunoblástico de células T.
Linfoma extranodal de células T y de células Nk, tipo nasal.
Linfoma intestinal de células T, de tipo enteropático.
Linfoma y leucemia de células T en adultos (HTLV 1+).
Linfoma anaplásico de células grandes, tipo sistémica primario.
Linfoma anaplásico de células grandes, tipo cutáneo primario.
Leucemia agresiva de células NK
Linfoma de Hodgkin (Enfermedad de Hodgkin)
Linfoma de Hodgkin nodular abundante en linfocitos.
Linfoma de Hodgkin clásico.
Linfoma de Hodgkin con esclerosis nodular.
Linfoma de Hodgkin clásico rico en linfocitos.
Linfoma de Hodgkin de celularidad mixta.
Linfoma de Hodgkin con depleción de linfocitos.
Linfomas de células asesinas naturales (NK) y células T periféricas.
Leucemia linfocítica y leucemia prolinfocítica crónicas de células T.
Leucemia linfocítica granular de células T.
Micosis fungoide y el síndrome de Sézary.
Linfoma periférico de célula T, sin alguna otra caracterización.
Linfoma hepatoesplénico de células T gamma y delta.
Linfoma de apariencia paniculítica subcutáneo de células T.
Linfoma angioinmunoblástico de células T.
Linfoma extranodal de células T y de células Nk, tipo nasal.
Linfoma intestinal de células T, de tipo enteropático.
Linfoma y leucemia de células T en adultos (HTLV 1+).
Linfoma anaplásico de células grandes, tipo sistémica primario.
Linfoma anaplásico de células grandes, tipo cutáneo primario.
Leucemia agresiva de células NK
Linfoma de Hodgkin (Enfermedad de Hodgkin)
Linfoma de Hodgkin nodular abundante en linfocitos.
Linfoma de Hodgkin clásico.
Linfoma de Hodgkin con esclerosis nodular.
Linfoma de Hodgkin clásico rico en linfocitos.
Linfoma de Hodgkin de celularidad mixta.
Linfoma de Hodgkin con depleción de linfocitos.

Estadificación de los linfomas
Se utiliza la clasificación de Ann Arbor para la Enfermedad de Hodgkin de 1971. Para poder estadiar un linfoma se precisa de información de la historia clínica, exploración física, técnicas de diagnóstico por la imagen, análisis de sangre, informe de la biopsia inicial y de la médula ósea.
*Estadios clínicos *
Estadio I: Afectación de una sola región ganglionar, o afectación localizada de un solo órgano o localización extralinfática.
Estadio II: Afectación de dos o más regiones ganglionares del mismo lado del diafragma, o afectación localizada de un solo órgano o localización extralinfática (E) y su ganglio o ganglios regionales con o sin afectación de otras regiones ganglionares en el mismo lado del diafragma.
Estadio III: Afectación de regiones ganglionares a ambos lados del diafragma, que puede acompañarse también de afectación localizada de un órgano o localización extralinfática asociada, o de afectación de bazo (S) o ambas (E+S).
Estadio IV: Afectación diseminada de uno o más órganos extralinfáticos, con o sin afectación ganglionar asociada, o afectación extralinfática aislada con afectación ganglionar a distancia. La afectación de médula ósea implica un estadio IV.
*Síntomas A y B*
Cada estadio clínico debe clasificarse en A y B dependiendo de la ausencia (A) o presencia (B) de síntomas generales definidos. Estos síntomas B son los siguientes.
Pérdida de peso inexplicada de más del 10% del peso corporal habitual en los últimos seis meses a la primera consulta médica.
Fiebre inexplicada con una temperatura superior a 38 grados centígrados. Una enfermedad febril breve asociada a una infección conocida no es un síntoma B.
Sudoración nocturna.



Regiones linfáticas y órganos extralinfáticos
Regiones linfáticas: Corresponden a localizaciones de ganglios linfáticos accesibles a la exploración física (palpación e inspección) como región cervicosupraclavicular, región axilar y región inguinal. Las regiones linfáticas sólo visualizadas por técnicas de imagen como el TAC son el mediastino, retroperitoneo y regiones mesentéricas. Existen estructuras linfáticas que también son consideradas como regiones linfáticas como el anillo de Waldeyer, bazo, apéndice, timo y placas de Peyer. Cuando se afecta al bazo se añade al estadio la letra S (del inglés spleen) y basta que esté aumentado de tamaño en la palpación o por técnicas de imagen, no siendo necesaria la biopsia esplénica.

Órganos extralinfáticos: Son los pulmones, hueso, hígado, cerebro, médula ósea, pleura, peritoneo, glándulas suprarrenales, piel y otros. La afectación hepática, aunque sea localizada, siempre se considera una afectación difusa.

*Tratamiento de los linfomas*
Cada linfoma tiene un tratamiento diferente, pero los tratamientos convencionales ahora incluyen diversos regímenes de quimioterapia, radioterapia, e inmunoterapia, o combinaciones de dichos tratamientos, dependiendo del paciente y su contexto.



.jpg)